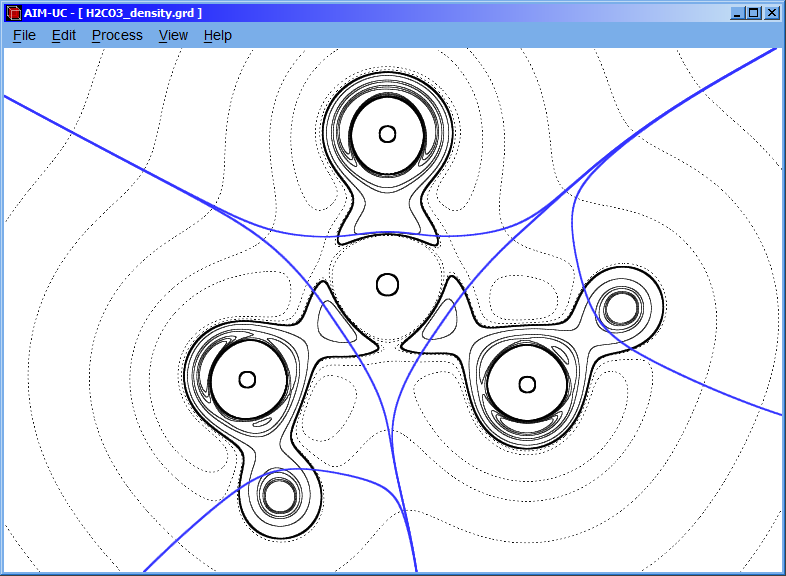
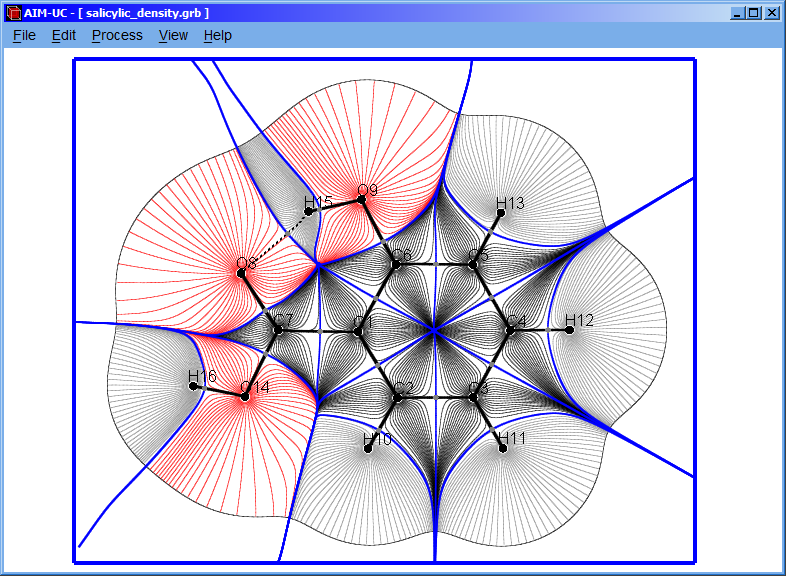
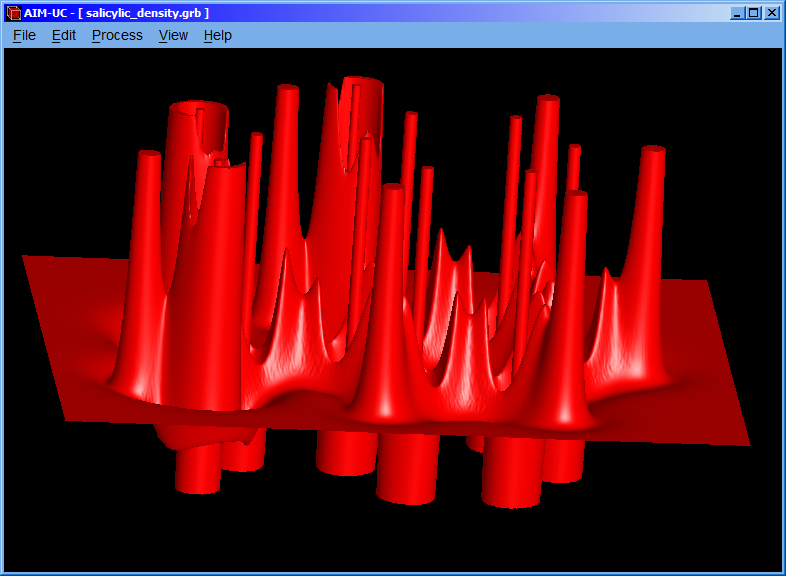
3.1 Calculating contour map
Click the “Process” option and then click the “Calculate Contour Map” option.
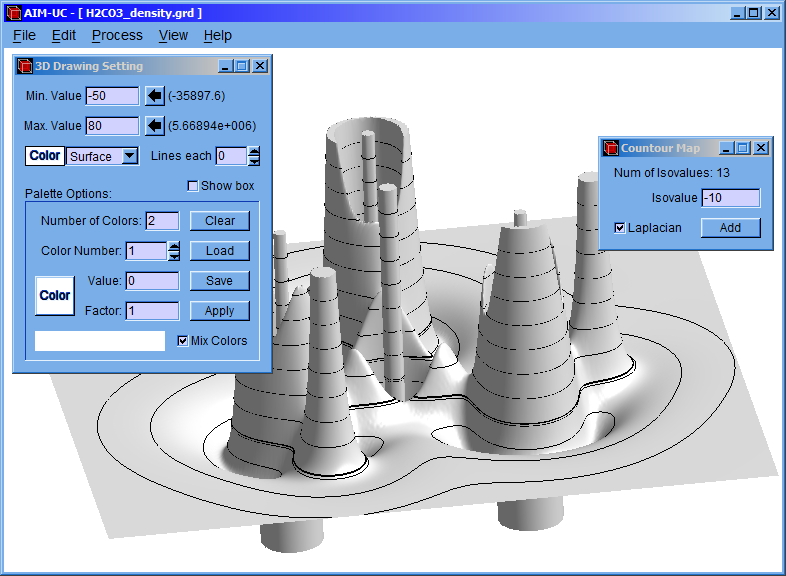
Following, a setting window will appear and introduce the isovalues of charge density. Then click the “add” button; if “Laplacian” checkbox is marked, then it will calculate the contour of the negative laplacian, instead of the contour of the charge density.
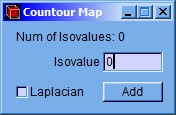
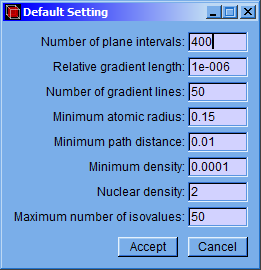
NOTE: The maximum number of isovalues that can be added is 50. This number can be modified in the “Default Settings” window, explained in section 5.
For viewing the contour map, click the “View” option and then click the “Change Display” option.
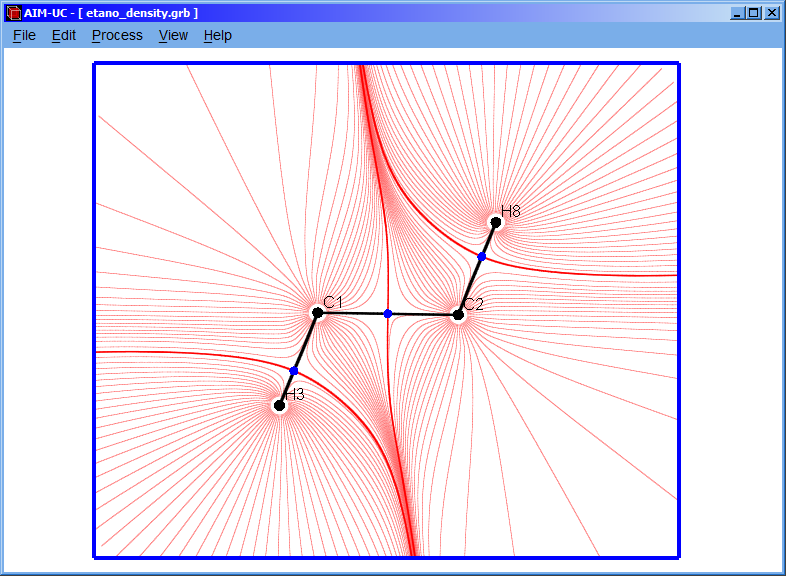
The contour line corresponding to the added isovalue can be visualized in this way.
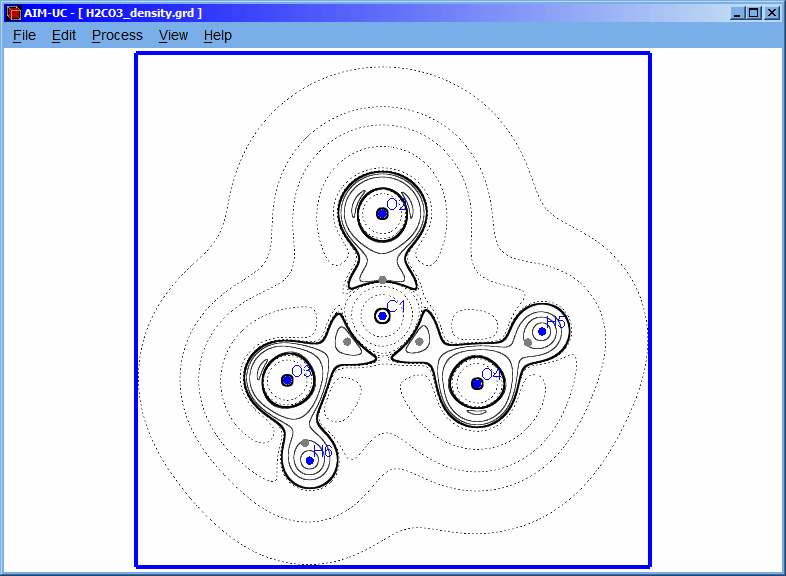
NOTE: If the plane dimensions or positions are changed, it is necessary to create again the plane and recalculate the contour map.
3.2 Finding plane critical points
The program finds the critical points in the 3D space contained by limit box and the charge density plane.
For calculating the critical points in the 3D grid, click the “Process” option and then click the “Find 3D Grid Critical Points” option. The critical points on the plane can be calculated by clicking the “Process” and the “Find Plane Critical Points” options.
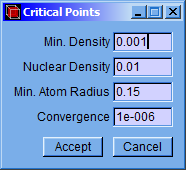
After that, a setting window (Critical Points) will appear allowing to modify minimum density, nuclear density, minimum atomic radius and convergence factor values. Although the program has default values, they can be easily changed in this window. Then click the “Accept” button to find the critical points.
-
Min. Density: Minimum value of the electronic charge density. A critical point with charge density below this value will not be stored.
Nuclear Density: Value of the electronic charge density; the search of maximum critical points will start up to this value. The high electronic charge density around the atom nuclei makes the gradient module increases too fast, under such a condition the Newton-Raphson method fails. Due to this fact a simple searching method is used for those points of the grid that have a charge density above the “Nuclear Density” value.
Min. Atom Radius: If the distance from an atom nucleus to a critical point is lower than this value, then the critical point will not be stored. This prevents fake critical points originated by the oscillation of the interpolation polynomial near the atom nuclei.
Convergence: Criterion applied to the gradient that allows deciding whether it is a critical point. In order to consider it as a critical point, the module of the gradient vector divided by the charge density, all this multiplied by itself must be lower than the Convergence value.
Information of the 3D critical points could be saved in a file. Click the “File” option followed by the “Save Critical Points” option.
NOTE: In the case of search of critical points (3D) in *.wfn files, the “Nuclear Density” value is not used. The searches start at the midpoints of every pair, trio and quartet of atoms.
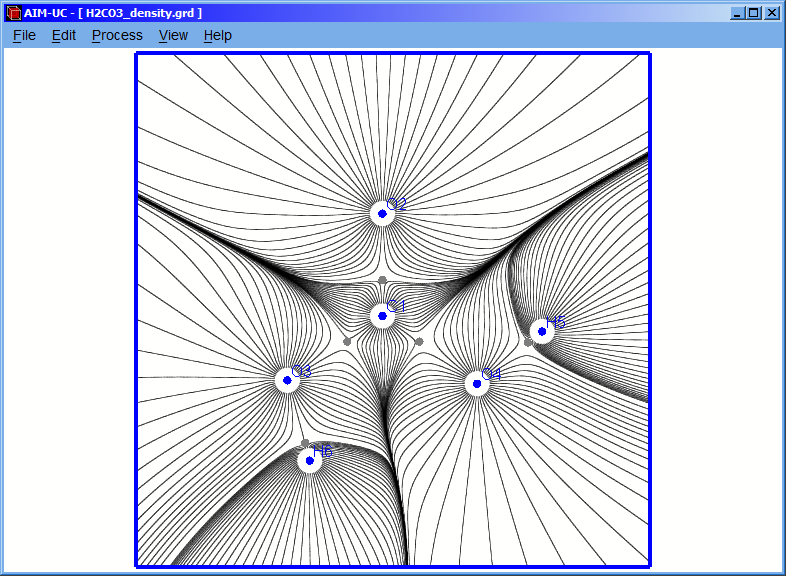
3.3 Calculating gradient field
Click the “Process” option and then the “Calculate Gradient Field” option.
NOTE: Plane critical points must be calculated before doing the gradient field calculation.
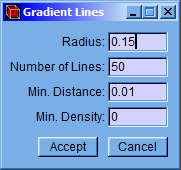
A setting window (Gradient Lines) will appear in which the values of the radius, number of gradient lines and the minimum distance can be set. Although the program has default values, they can be easily changed in this window. Then click “Accept” button to calculate the gradient field.
-
Radius: Distance from the nucleus to the beginning point of the gradient lines.
Number of lines: Amount of gradient lines that will be shown around each atom.
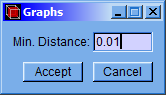
Min. Distance: Minimum distance between two consecutive points of the gradient path (The fourth order Runge-Kutta method is used, and the path follows the negative gradient).
Min. Density: The electronic charge density is evaluated in each point of the gradient path; the calculation of the current path stops if it is less than the “Min. Density” value.
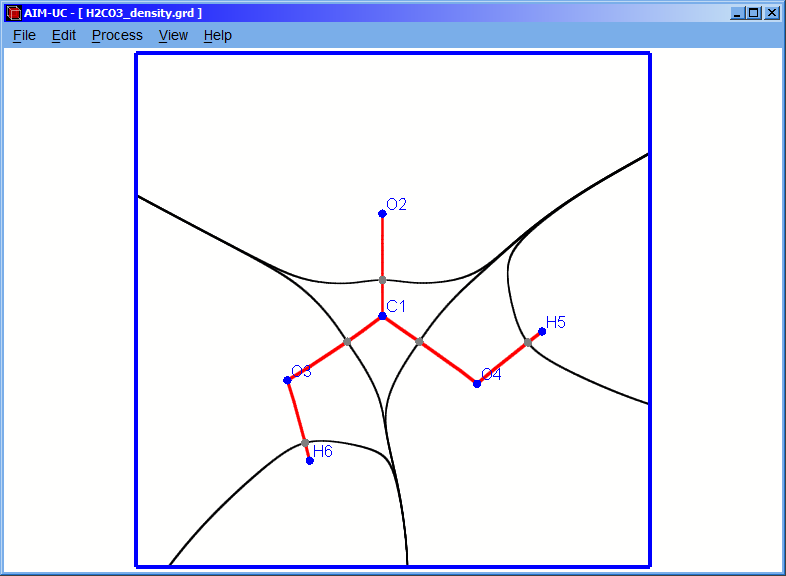
3.4 Calculating bond and basin paths
Click the “Process” option and then the “Calculate Bond and Basin Paths” option.
A setting window will appear in which the minimum distance value can be modified, then click the “Accept” button.
NOTE: Before calculating the bond and basin paths, the plane critical points must be found.
NOTE: It is also recommendable to calculate the 3D critical points (explained in section 3.2) to correct the molecular graph, hiding the paths that do not represent any bond.
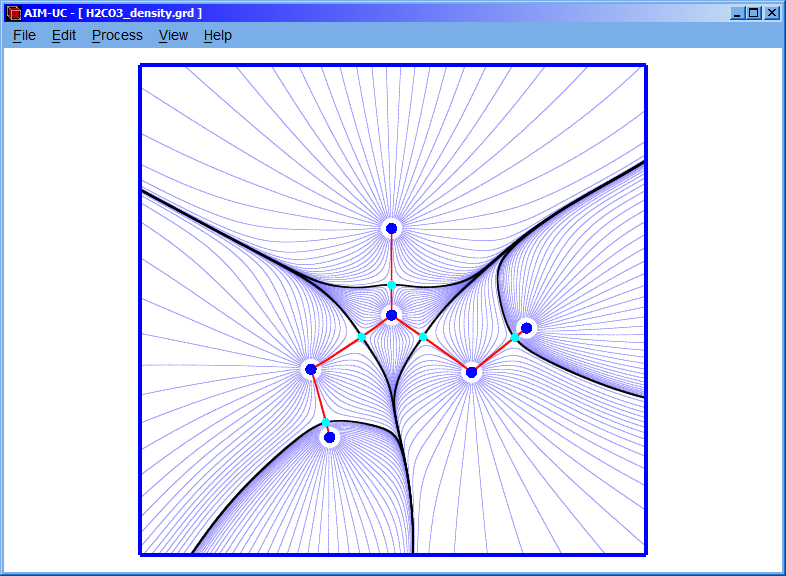
3.5 Processing all ¤ items
By clicking on an option it is possible to calculate the plane grid (if it was not calculated), the critical points, the gradient field, the molecular graph and the interatomic basin bounds. For this, click menu “Process” option and then the “Processing all ¤ item” option, after a few seconds an information window will appear indicating that all those items were successfully calculated.
3.6 2D Drawing settings
Click the menu “Edit” option and then the “2D Drawing Settings” option.
A setting window with tabs will appear allowing modify some characteristics of the drawing.
3.6.1 General settings
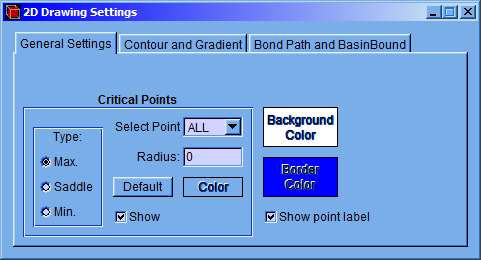
In the “General Settings” tab of the “2D Drawing Settings” window the “Background Color” and the “Border Color” can be selected. The “Show point label” checkbox will show the critical point tags; the “Radius” and “Color” of the different critical points on the plane: maximum (Max.), minimum (Min.) and saddle points (Saddle) can be modified. Besides, the “Show” checkbox allows whether a critical point will be shown.
3.6.2 Modifying contour map and gradient lines
In the “Contour and Gradient” tab some characteristics of the contour map and the gradient field can be modified.
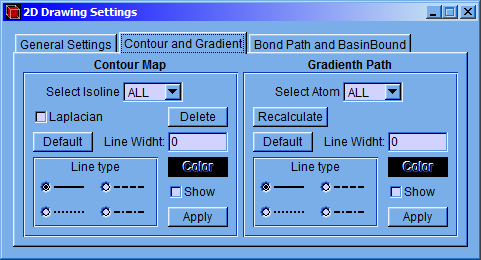
On the left side of the window, the “Contour Map” section, the type, color, and width of the line can be changed and deciding whether the line is shown. These modifications can be applied to individual isoline (selecting just one isovalue in “Select Isoline”) or to the group (selecting “ALL”). The characteristics of the lines can be turned back to their defaults by pressing the “Default” button.
After modify the characteristics, click the “Apply” button to enter the changes.
NOTE: When the “Laplacian” checkbox is activated, the characteristics of the negative laplacian contour map (instead of the density contour map) will be modified.
On the right side of the window, the “Gradient Path” section, the characteristics of the gradient field lines that belong to any atom can be modified. Clicking the “Select Atom” menu the gradient lines of a specific atom or all of them can be modified. Click the “Apply” button to enter the changes.
By clicking the “Recalculate” button, it is possible to change all the options explained in section 3.3, for a single atom or all of them.
3.6.3 Modifying molecular graph and interatomic basin bounds
In the “Bond Path and BasinBound” tab the characteristics of the molecular graph and interatomic basin bounds can be modified.
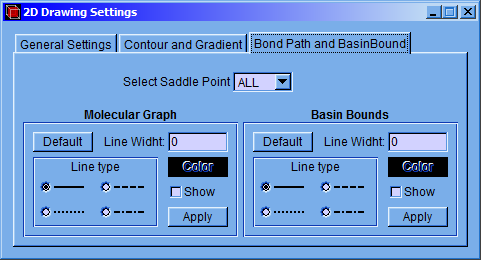
On the left side of the window, “Molecular Graph” section, the type, color, and width of the line can be set and deciding whether the line is shown. On the right side, “Basin Bounds” section, it is possible to do the above indicated changes with the lines of the interatomic basin bound. These modifications can be applied to individual or group by selecting a single saddle point or all of them in the “Select Saddle Point” menu. The characteristics of the lines can be turned back to their defaults by pressing the “Default” button.
[Table of Contents]